 中文网站
中文网站
NEJM julkaisi verkossa 29. joulukuuta aamuyöllä uuden kliinisen vaiheen III tutkimuksen uudesta kiinalaisesta koronaviruksesta VV116. Tulokset osoittivat, että VV116 ei ollut kliinisen toipumisen keston suhteen huonompi kuin Paxlovid (nematoviiri/ritonaviiri) ja että sillä oli vähemmän haittavaikutuksia.

Kuvan lähde: NEJM
Mediaani toipumisaika 4 päivää, haittavaikutusten määrä 67,4 %
VV116 on suun kautta otettava nukleosidilääke uuden koronaviruksen (SARS-CoV-2) hoitoon, joka on kehitetty yhteistyössä Junsitin ja Wang Shan Wang Shuin kanssa. Se on RdRp-estäjä yhdessä Gileadin remdesiviirin, Merck Sharp & Dohmen molnupiraviirin ja Real Biologicsin atselvudiinin kanssa.
Vuonna 2021 Uzbekistanissa saatiin päätökseen VV116-valmisteen faasi II kliininen tutkimus. Tutkimuksen tulokset osoittivat, että VV116-ryhmä pystyi parantamaan kliinisiä oireita paremmin ja vähentämään merkittävästi kriittisen vaiheen etenemisen ja kuoleman riskiä verrattuna kontrolliryhmään. Tutkimuksen positiivisten tulosten perusteella VV116 on hyväksytty Uzbekistanissa keskivaikean tai vaikean COVID-19-taudin hoitoon, ja siitä on tullut ensimmäinen uusi suun kautta otettava sepelvaltimolääke, joka on hyväksytty markkinoitavaksi ulkomailla Kiinassa [1].
Tämä vaiheen III kliininen tutkimus[2] (NCT05341609), jota johtivat professori Zhao Ren Shanghain Ruijin-sairaalasta, professori Gaoyuan Shanghain Renji-sairaalasta ja akateemikko Ning Guang Shanghain Ruijin-sairaalasta, saatiin päätökseen Omicron-variantin (B.1.1.529) aiheuttaman epidemian aikana maalis-toukokuussa Shanghaissa. Tutkimuksen tavoitteena oli arvioida VV116:n tehoa ja turvallisuutta Paxlovidiin verrattuna lievää tai kohtalaista COVID-19-tautia sairastavien potilaiden varhaisessa hoidossa. Tavoitteena oli arvioida VV116:n tehoa ja turvallisuutta Paxlovidiin verrattuna lievää tai kohtalaista COVID-19-tautia sairastavien potilaiden varhaisessa hoidossa.

Kuvalähde: Viite 2
Monikeskustutkimuksessa, jossa ei havaittu havaintoja, tutkijat olivat sokkoutettuja ja kontrolloituja, tutkittiin 822 aikuista Covid-19-potilasta, joilla oli korkea taudin etenemisriski ja lieviä tai kohtalaisia oireita. Tutkimukseen osallistujien kelpoisuutta arvioitiin seitsemästä Shanghain, Kiinan, sairaalasta. Lopulta 771 osallistujaa sai joko VV116-lääkettä (384, 600 mg 12 tunnin välein päivänä 1 ja 300 mg 12 tunnin välein päivinä 2–5) tai Paxovidia (387, 300 mg nimatuviiria + 100 mg ritonaviiria 12 tunnin välein 5 päivän ajan) suun kautta otettavana lääkkeenä.
Tämän kliinisen tutkimuksen tulokset osoittivat, että varhainen VV116-hoito lievässä tai kohtalaisessa COVID-19-taudissa saavutti kliinisen protokollan ennustaman ensisijaisen päätetapahtuman (aika pysyvään kliiniseen toipumiseen): mediaaniaika kliiniseen toipumiseen oli 4 päivää VV116-ryhmässä ja 5 päivää Paxlovid-ryhmässä (riskisuhde 1,17; 95 %:n luottamusväli 1,02–1,36; alaraja >0,8).
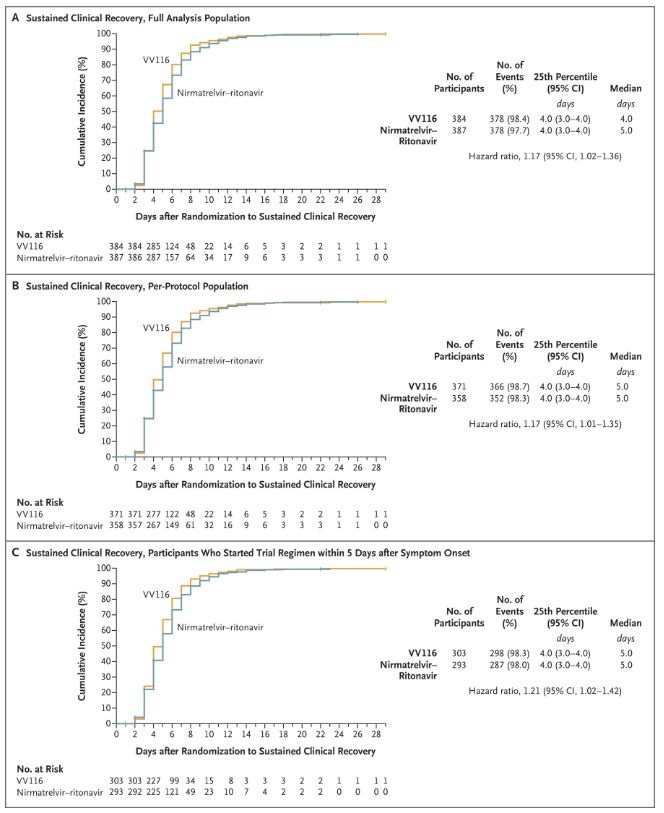
Kliinisen toipumisajan ylläpitäminen
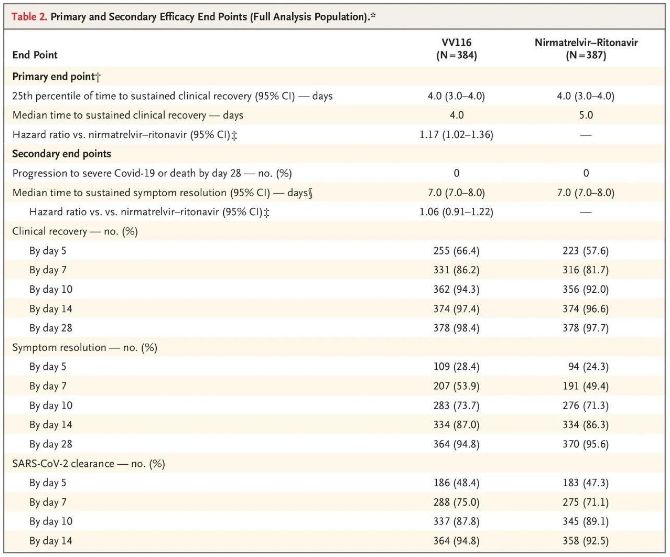
Ensisijaiset ja toissijaiset tehon päätetapahtumat (kokonaispopulaatioanalyysi)
Kuvalähde: Viite 2
Turvallisuuden osalta VV116-rokotetta saaneet osallistujat raportoivat vähemmän haittatapahtumia (67,4 %) kuin Paxlovidia saaneet (77,3 %) 28 päivän seurannassa, ja asteen 3/4 haittatapahtumien ilmaantuvuus oli VV116-ryhmässä pienempi (2,6 %) kuin Paxlovid-ryhmässä (5,7 %).
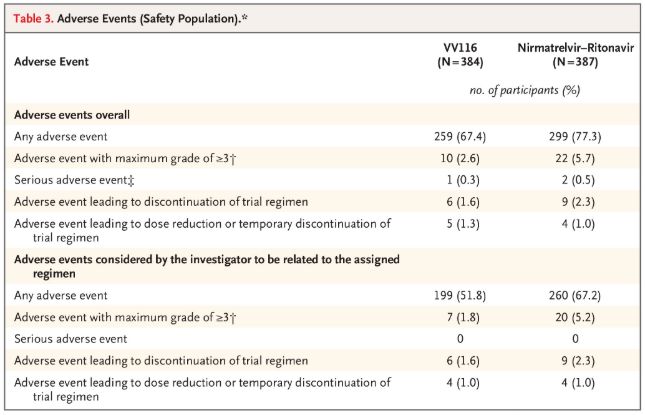
Haittatapahtumat (turvalliset ihmiset)
Kuvalähde: Viite 2
Kiistat ja kysymykset
Juniper ilmoitti 23. toukokuuta 2022, että VV116:n ja PAXLOVIDin vertailu kliinisessä vaiheen III rekisteröintitutkimuksessa lievän tai kohtalaisen COVID-19:n varhaisessa hoidossa (NCT05341609) saavutettiin tutkimuksen ensisijainen päätetapahtuma.

Kuvalähde: Viite 1
Aikana, jolloin tutkimuksesta puuttui yksityiskohtia, vaiheen III tutkimusta ympäröivä kiista oli kaksijakoinen: ensinnäkin se oli yksisokkoutettu tutkimus, ja lumekontrollin puuttuessa pelättiin, että lääkkeen täysin objektiivinen arviointi olisi vaikeaa; toiseksi kliinisistä päätetapahtumista oli epäilyksiä.
Juniperin kliiniset sisäänottokriteerit ovat (i) positiiviset tulokset uudessa koronatestissä, (ii) yksi tai useampi lievä tai kohtalainen COVID-19-oire ja (iii) potilaat, joilla on suuri riski saada vakava COVID-19, mukaan lukien kuolema. Ainoa ensisijainen kliininen päätetapahtuma on kuitenkin "aika kestävään kliiniseen toipumiseen".
Juuri ennen ilmoitusta, 14. toukokuuta, Juniper oli tarkistanut kliinisiä päätetapahtumia poistamalla yhden kliinisistä ensisijaisista päätetapahtumista, "vakavaan sairauteen tai kuolemaan johtavien konversioiden osuuden" [3].
![]()
Kuvalähde: Viite 1
Näitä kahta keskeistä kiistakohtaa käsiteltiin myös julkaistussa tutkimuksessa erikseen.
Omicronin äkillisen puhkeamisen vuoksi Paxlovidin lumelääketablettien tuotantoa ei ollut saatu valmiiksi ennen tutkimuksen alkua, joten tutkijat eivät voineet suorittaa tätä tutkimusta kaksoissokkoutetulla, kaksoismallimenetelmällä. Kliinisen tutkimuksen yksöissokkoutetun puolen osalta Juniper totesi, että protokolla toteutettiin sääntelyviranomaisten kanssa käydyn keskustelun jälkeen ja että yksöissokkoutettu suunnittelu tarkoittaa, että tutkija (mukaan lukien tutkimuksen päätetapahtuman arvioija) tai sponsori eivät tiedä tarkkaa terapeuttista lääkeainejakoa ennen kuin lopullinen tietokanta lukitaan tutkimuksen lopussa.
Lopulliseen analyysiin mennessä kukaan tutkimukseen osallistuneista ei ollut kuollut tai edennyt vakavaan Covid-19-tapahtumaan, joten VV116:n tehosta vaikean tai kriittisen Covid-19-taudin etenemisen tai kuoleman ehkäisyssä ei voida tehdä johtopäätöksiä. Tiedot osoittivat, että arvioitu mediaaniaika satunnaistamisesta Covid-19:ään liittyvien kohdeoireiden pysyvään regressioon oli 7 päivää (95 %:n luottamusväli 7–8) molemmissa ryhmissä (riskisuhde 1,06; 95 %:n luottamusväli 0,91–1,22) [2]. Ei ole vaikea selittää, miksi ensisijainen päätetapahtuma, "vakavaan sairauteen tai kuolemaan johtava nopeus", joka alun perin asetettiin ennen tutkimuksen päättymistä, poistettiin.
Emerging Microbes & Infections -lehti julkaisi 18. toukokuuta 2022 ensimmäisen kliinisen VV116-tutkimuksen tulokset Omicron-variantilla infektoituneilla potilailla [4]. Kyseessä oli avoin, prospektiivinen kohorttitutkimus, johon osallistui 136 vahvistettua sairaalahoitopotilasta.
Tutkimuksen tiedot osoittivat, että Omicron-infektiota sairastavilla potilailla, jotka käyttivät VV116-valmistetta viiden päivän kuluessa ensimmäisestä positiivisesta nukleiinihappotestistä, nukleiinihappojen regressioaika oli 8,56 päivää, mikä on vähemmän kuin kontrolliryhmässä, jossa vastaava aika oli 11,13 päivää. VV116:n anto oireileville potilaille tämän tutkimuksen aikataulun mukaisesti (2–10 päivää ensimmäisestä positiivisesta nukleiinihappotestistä) lyhensi nukleiinihappojen regressioaikaa kaikilla potilailla. Lääketurvallisuuden osalta VV116-hoitoryhmässä ei havaittu vakavia haittavaikutuksia.
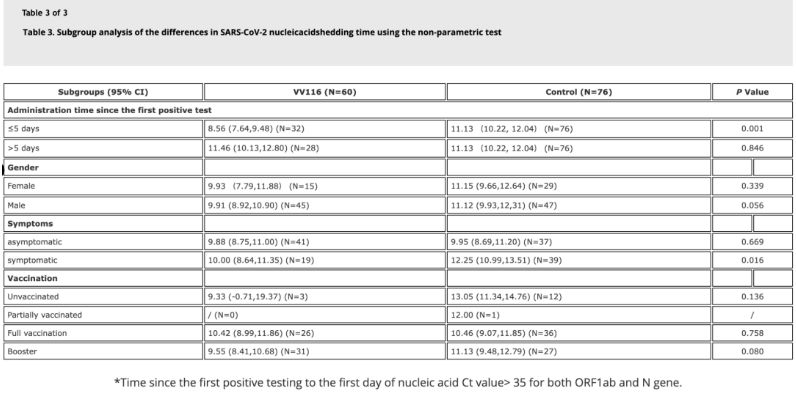
Kuvalähde: Viite 4
VV116-valmisteella on meneillään kolme kliinistä tutkimusta, joista kaksi on lievää tai kohtalaista COVID-19-tautia käsitteleviä faasi III -tutkimuksia (NCT05242042, NCT05582629). Toinen kohtalaista tai vaikeaa COVID-19-tautia koskeva tutkimus on kansainvälinen, monikeskustutkimus, satunnaistettu, kaksoissokkoutettu faasi III -kliininen tutkimus (NCT05279235), jossa arvioidaan VV116:n tehoa ja turvallisuutta verrattuna standardihoitoon. Juniperin ilmoituksen mukaan ensimmäinen potilas otettiin mukaan tutkimukseen ja hänelle annettiin lääke maaliskuussa 2022.

Kuvalähde: clinicaltrials.gov
Viitteet:
[1]Junshi Biotech: Ilmoitus VV116:n ja PAXLOVIDin vertailua koskevan vaiheen III rekisteröidyn kliinisen tutkimuksen pääasiallisesta päätetapahtumasta lievän tai kohtalaisen COVID-19:n varhaisessa hoidossa
2 Xu, Hao Yin, Zhiren Fu, Hao Xing, Li Li, Liying Sun, Heyu Huang, Quanbao Zhang, Linlin Xu, Yanting Jin, Rui Chen, Guoyue Lv, Zhijun Zhu, Wenhong Zhang, Zhengxin Wang. (2022) Omicron-infektioprofiili ja rokotustila 1881 maksansiirron saajan keskuudessa: monikeskusinen retrospektiivinen kohortti. Emerging Microbes & Infections 11:1, sivut 2636-2644.
Julkaisun aika: 06.01.2023
 Tietosuoja-asetukset
Tietosuoja-asetukset